
type
status
date
slug
summary
tags
category
icon
password
Property
Mar 6, 2024 11:23 AM
Created time
Jan 31, 2024 05:37 PM
本文是《数据挖掘》课程的期中作业
一、实验目的
了解主成分分析的目的,内容以及流程。掌握主成分分析,编程实现癌症数据集的主成分分析,针对数据集中的癌症类型实现主成分下的数据可视化比较分析。
掌握类特征化和类对比分析,实现基于信息增益的属性相关分析,针对数据集中任意三种癌症数据实现癌症类概念描述及类特征化分析,并进行可视化。
二、实验过程及结果分析
主成分分析
2.1.1算法原理
主成分分析的目的是减少数据集变量数量,同时要保留尽可能多的特征信息;方法是通过正交变换将原始变量组转换为数量较少的彼此独立的特征变量,从而减少数据集的维数。
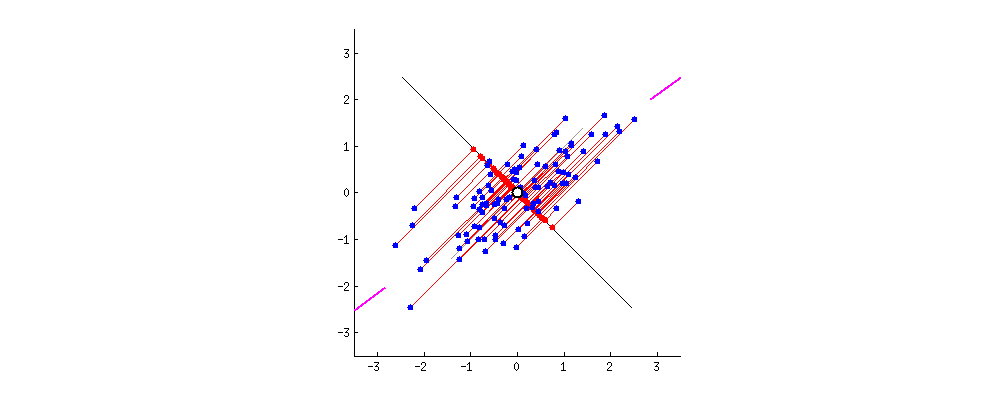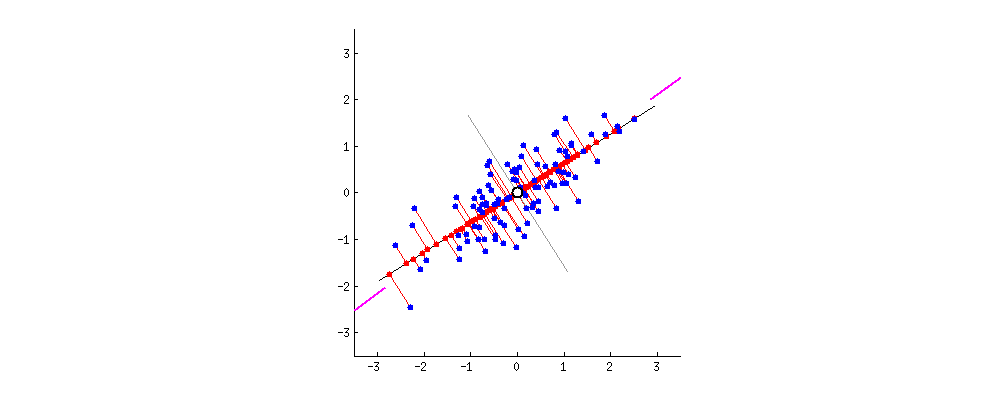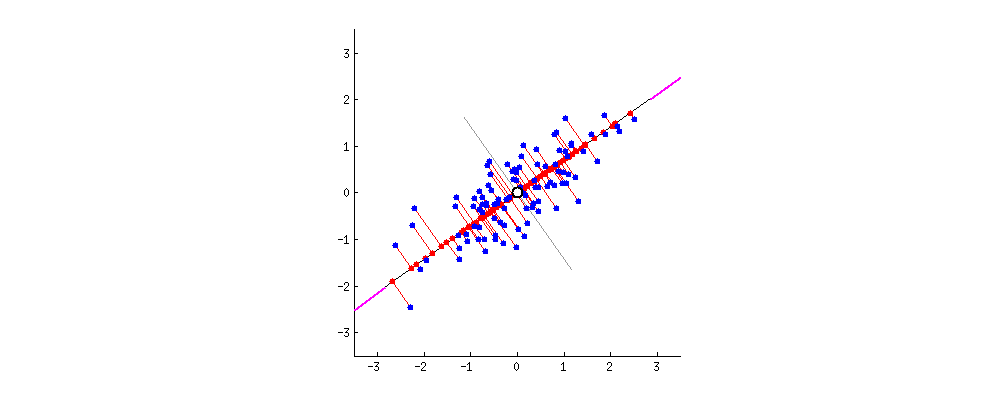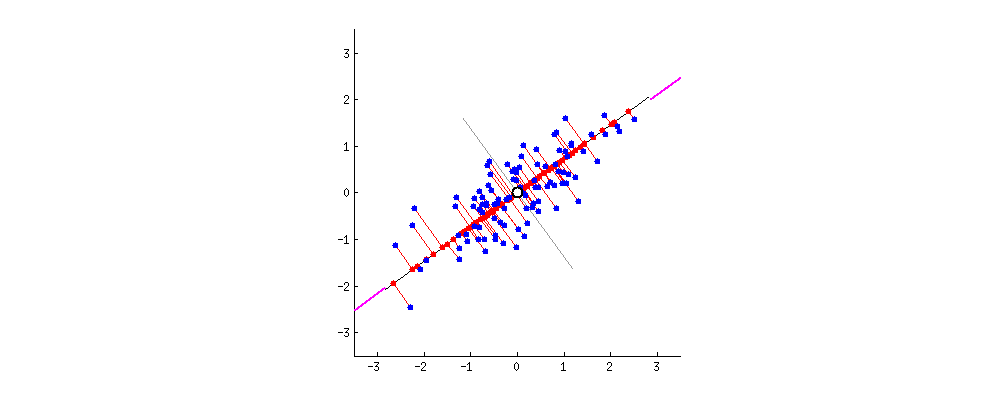
主成分分析方法的思想是,将高维特征(n维)映射到低维空间(k维)上,新的低维特征是在原有的高维特征基础上通过线性组合而重构的,并具有相互正交的特性,即为主成分。
通过正交变换构造彼此正交的新的特征向量,这些特征向量组成了新的特征空间。将特征向量按特征值排序后,样本数据集中所包含的全部方差,大部分就包含在前几个特征向量中,其后的特征向量所含的方差很小。因此,可以只保留前 k个特征向量,而忽略其它的特征向量,实现对数据特征的降维处理。
2.1.2 算法步骤
- 归一化处理,数据减去平均值;
- 通过特征值分解,计算协方差矩阵;
- 计算协方差矩阵的特征值和特征向量;
- 将特征值从大到小排序;
- 依次选取特征值最大的k个特征向量作为主成分,直到其累计方差贡献率达到要求;
- 将原始数据映射到选取的主成分空间,得到降维后的数据。
2.1.2算法实现
下方代码已有详细注释,不做更多说明。
2.1.3数据导入
本数据集的样本标签(如“TCGA-HQ-A2OF”)的元数据是不完整的,本来该标签应当续接该样本的类型,即到底是肿瘤样本还是正常对照,这里缺失了。
而老师未提供更多的数据说明,故这里认为这些样本都是肿瘤样本。
下面是数据的读取整合代码。
2.1.4 主成分分析
2.1.5 可视化处理
为结果还原一些标签,如该数据对应的癌症类型,方便后续的可视化处理
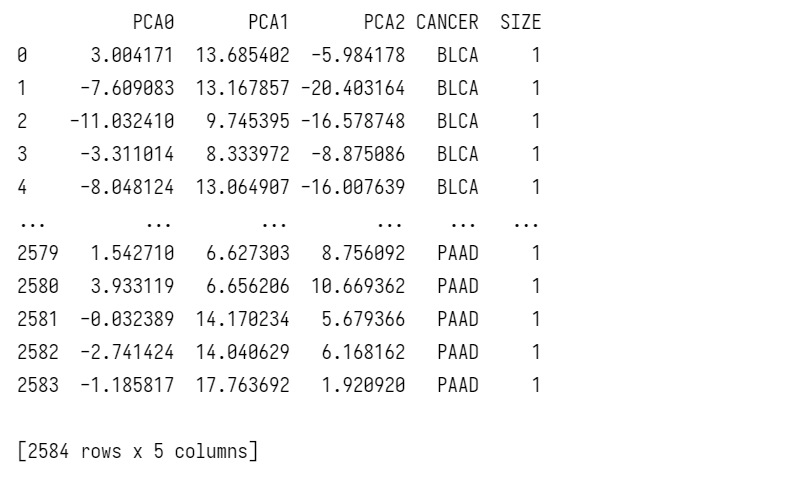
下方代码用于绘制95%置信椭圆,代码参考自https://gist.github.com/dpfoose/38ca2f5aee2aea175ecc6e599ca6e973
绘制二维图表
绘制三维图表
绘制方差累计贡献率
2.1.6 实验结果
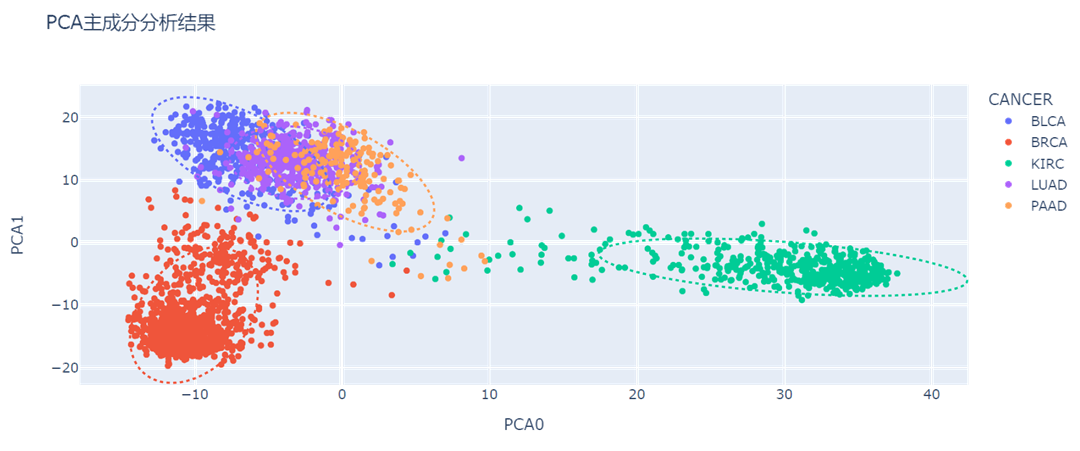
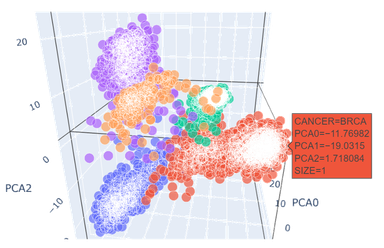
- 可以看到,只取2个主成分时,BRCA(乳腺浸润癌,上图红色),KIRC(肾透明细胞癌,上图绿色)与其他癌症样本的区分度较好;
- BLCA(膀胱尿路上皮癌,上图蓝色),LUAD(肺腺癌,上图紫色),PAAD(前列腺癌,上图橙色)的特征提取效果差,混杂度高,需要提高维度。
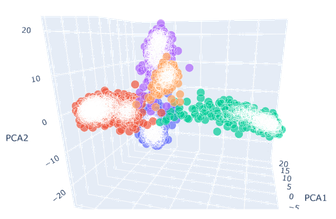
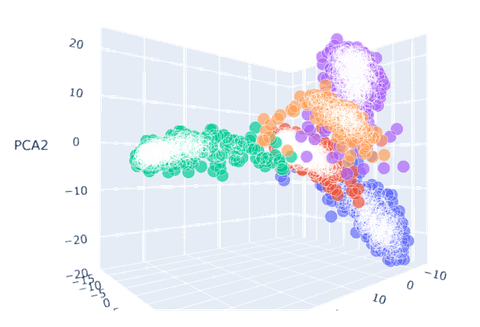
- 可以看到保留三个主成分后绘制的三维图像中,原先混杂度高的三种癌症的区分度都有了提升。其中以BLCA(膀胱尿路上皮癌,上图蓝色)的效果最好,LUAD(肺腺癌,上图紫色)和PAAD(前列腺癌,上图橙色)的效果次之。
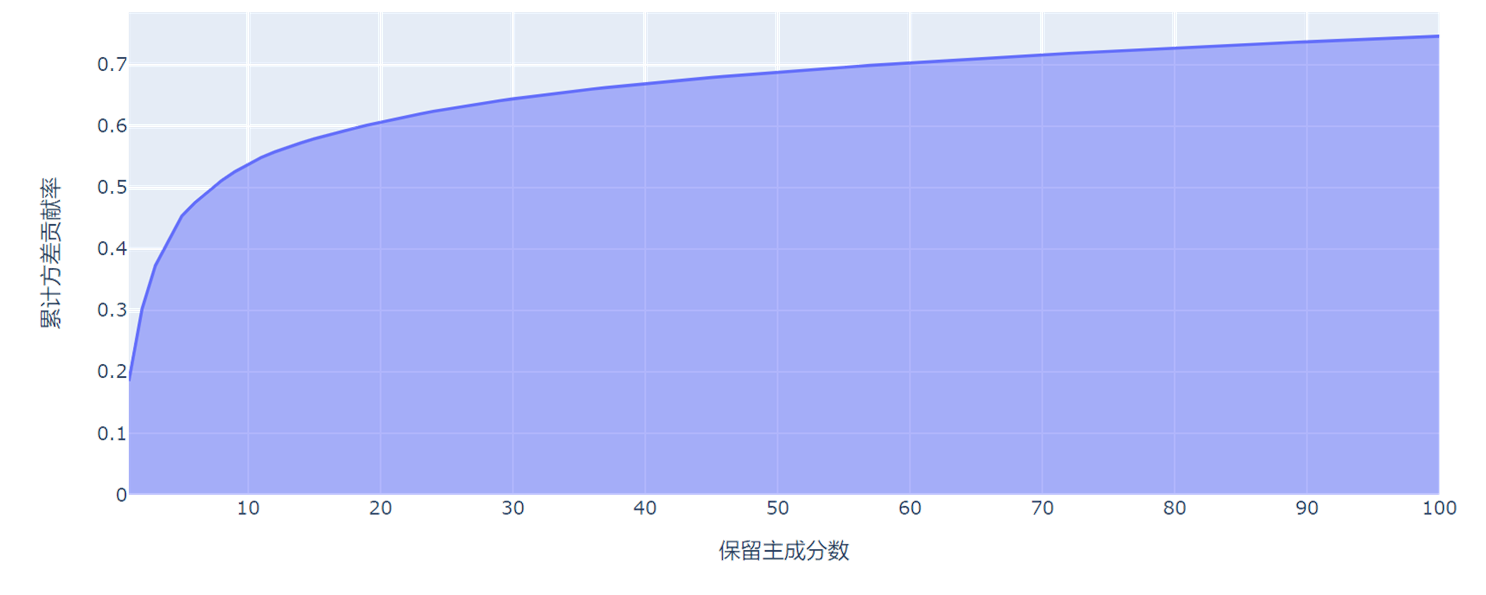
- 可以看到随着保留主成分数的增大,累计方差贡献率也不断提高,逐渐趋于1。同时,当保留数N=10时,累计方差也只占8%,考虑原因是本实验数据集的属性(基因)非常多,降维不能降得太低。当N=100时,累计方差贡献率方可达到74.6%。
2.2 类概念描述及特征化分析
基于属性归纳的数据概化方法由属性删除和属性概化完成。这里的属性均为基因,但是数据集没有更多的说明,我在基因功能与表达相似度的方面了解甚少,因此无法设计出更加有效的属性归纳算法。
本数据集中基因表达量的数值为连续值浮点数,考虑到后续的信息增益使用,这里需要对数据进行离散化处理。
在数据分箱方式的选择上我最开始选择了有监督的信息增益分箱,算法类似C4.5中的连续值分裂方法,但是发现该算法的递归深度以及计算复杂度特别高,计算一个特征就需要数十秒,本数据集有三千多个特征,因此这种方法效率过低就被舍弃了。因为实验要求使用信息增益,所以我就不考虑基于基尼系数的CART算法了。
2.2.1 属性概化-离散化
考虑到在同样的数据来源与测量设备下。不同基因的表达量量纲应当都是相同的,这里我绘制了所有基因表达量数据的频率直方图(3217*(399+1031+488)个数据)。
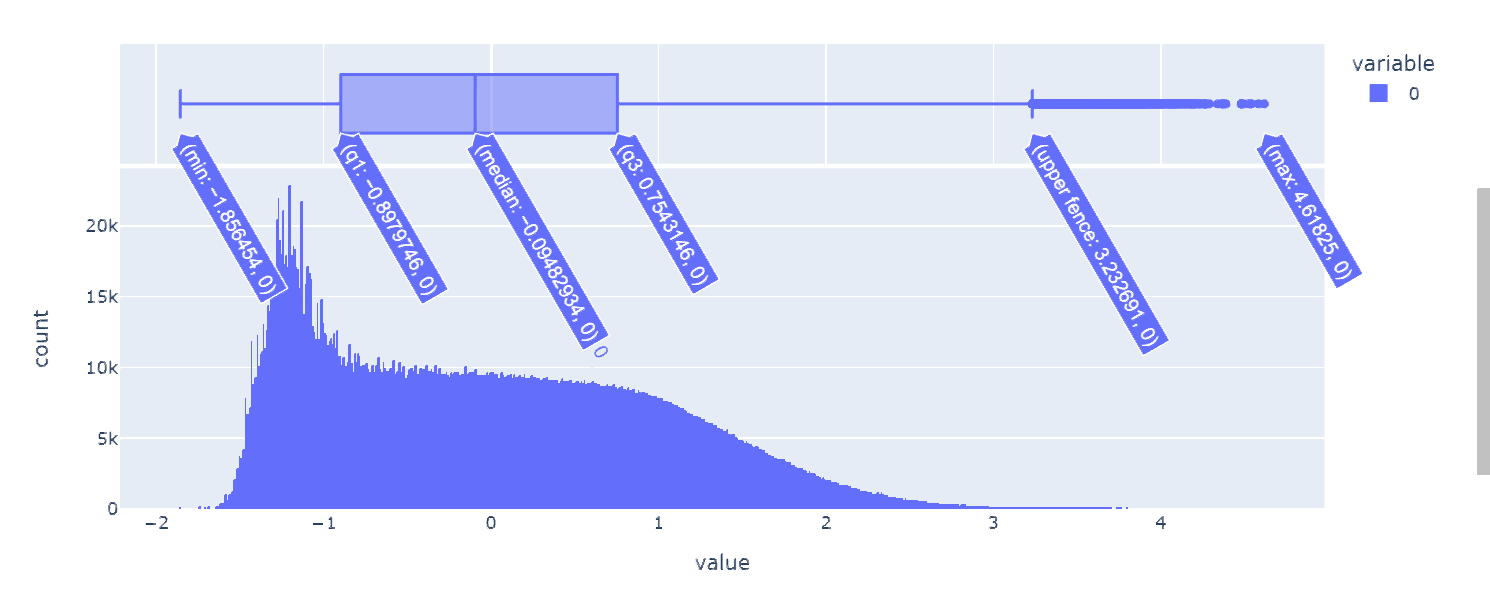
根据样本数据的频率分布,选择(-∞,-1],(-1,0],(0,1],(1,2],(2,+∞)来从小到大表征五种表达量高低程度,实现数据离散化,得到['极低', '低', '中', '高','极高']五种属性值,下面是离散化的实现代码。
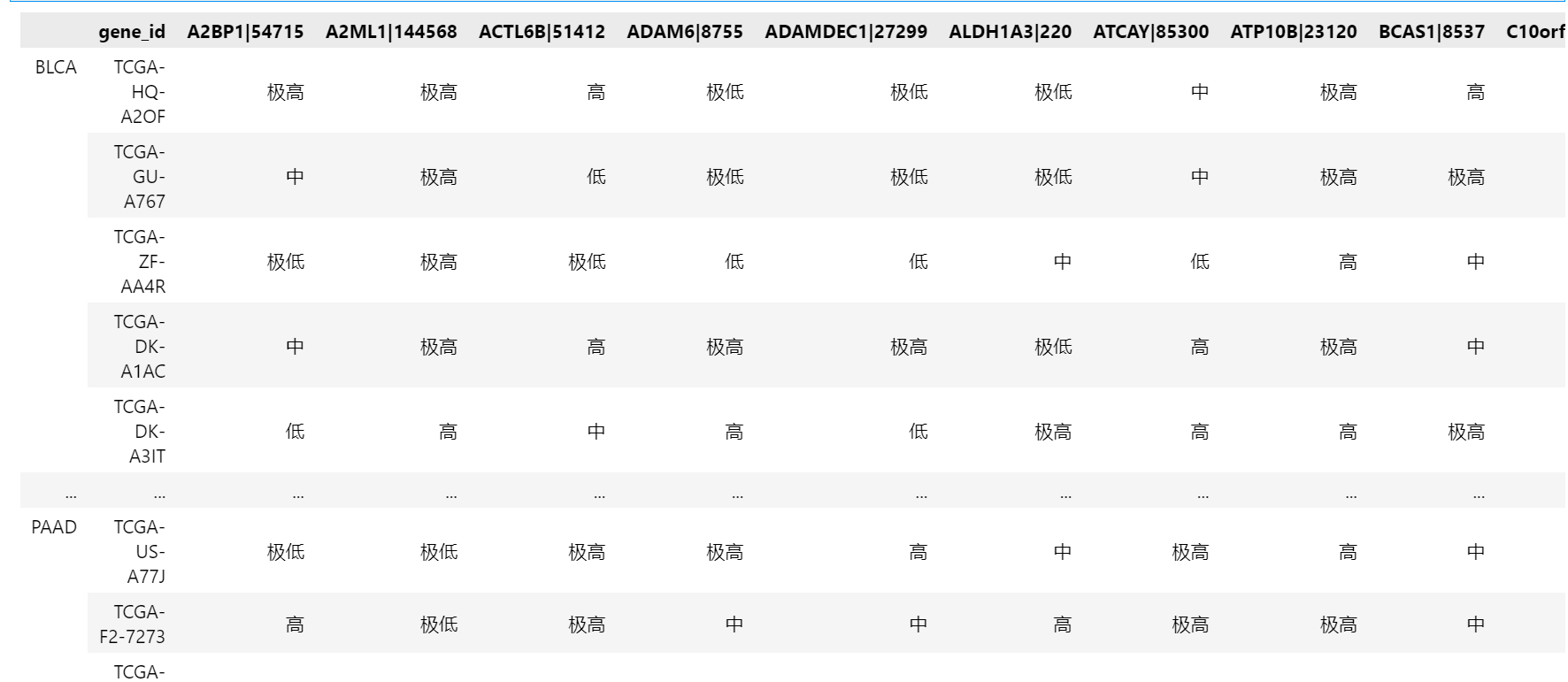
2.2.2 类特征化分析
这里按要求使用信息增益进行作为度量,进行属性相关分析下的类特征化分析。
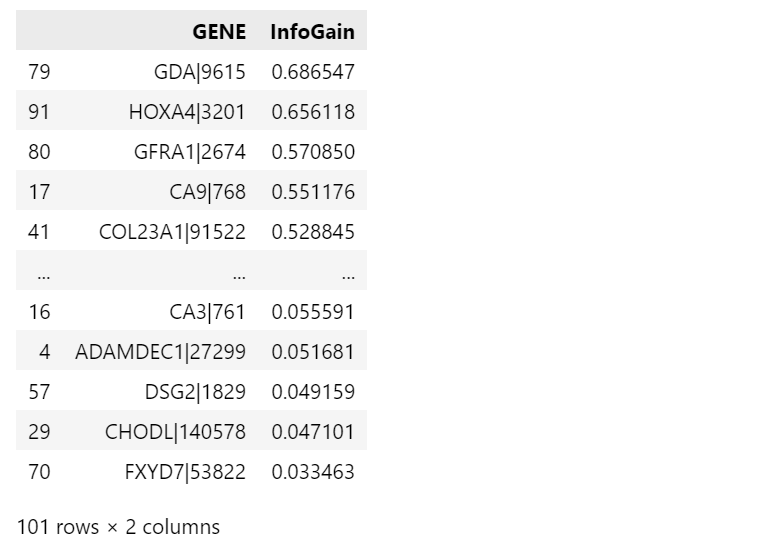
2.2.3 可视化分析
上述数据是以所有属性为根计算的信息增益结果,接下来将信息增益阈值设置为0.77,整理数据得到前10个特征进行可视化分析。
绘制多组箱型图进行可视化观察
2.2.4 类比较分析
选取信息增益排序的前三个属性,对所有概念元组进行计算d权,得到量化区分规则。
2.2.5 决策树的建立
在实验过程中考虑到只计算单轮信息增益的局限性,这里我试图借助ID3算法递归计算信息增益,按照决策树的返回结构得到被选出的特征属性,囿于时间关系,没有做更多的分析了。
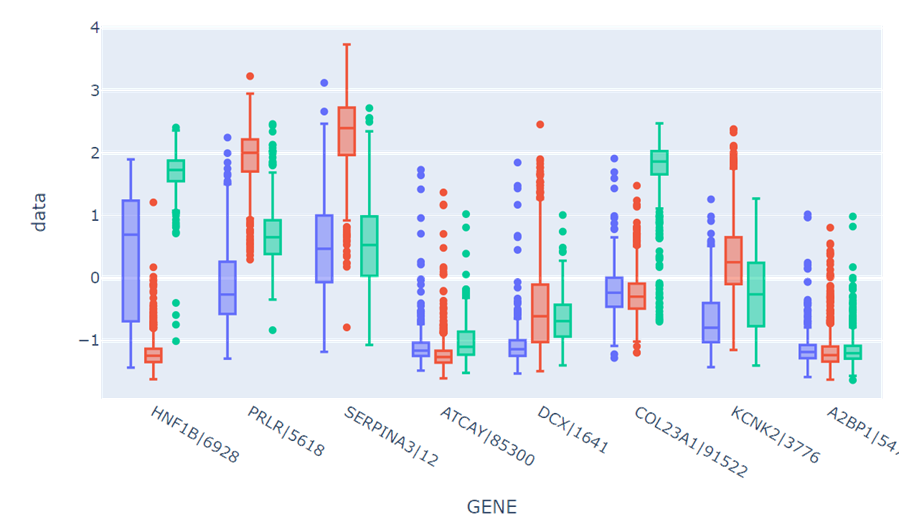

2.2.5类特征化结果分析

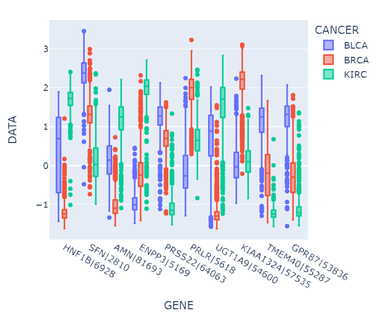
观察箱型图,可以发现提取出的特征在统计数值上有较好的区分度,即选出的基因在不同癌症类型下的表达量都有较大差异,能够较好地分开,说明这些基因与癌症标签的相关性强,因此属性相关分析的效果较好。
但是另一方面,上述信息增益的计算只考虑了各个属性与标签的相关性,忽视了属性间的相干性,例如第一个基因HNF1B|6928与第三个基因AMN|81693,两者都表现出BLCA下的中等水平表达,BRCA下的极低水平表达和KIRC下的高水平表达。因此属性之间的相干性很差,对此应当可以做相关系数热图分析进行属性约简。
2.2.6类比较结果分析
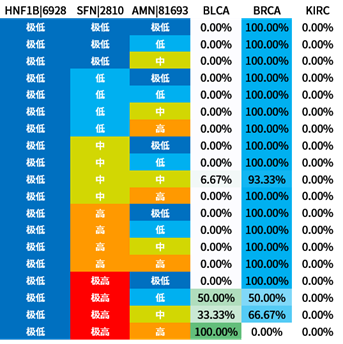
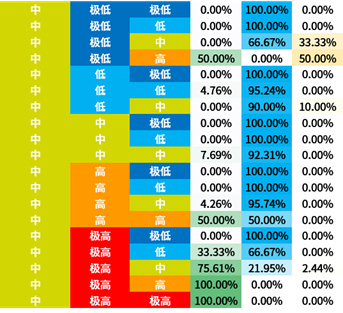
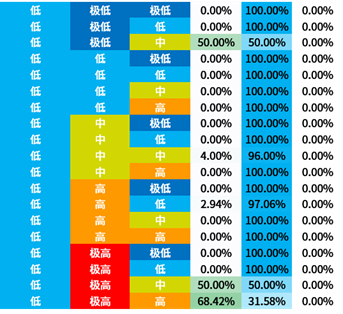
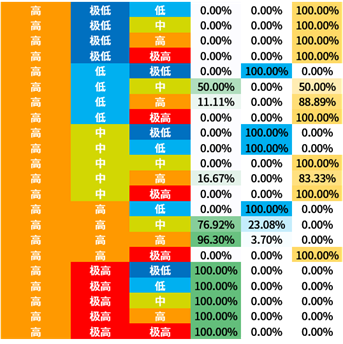
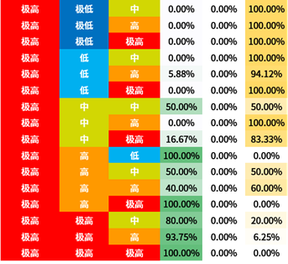
量化区分规则表也容易观察出:
- BLCA与2号基因极高表达量的关系显著
- BRCA与1号基因中低表达量的关系显著
- KIRC与1号基因高表达量的关系显著
3号基因因为排序不整齐所以不容易观察。
三、实验总结
本次实验总体完成了实验目标,实验结果符合预期,可视化输出效果较好。不仅加深了对数据挖掘相关技术的理解,也加强了我对Python下多种数据处理工具的运用能力。
- 作者:叶修齐
- 链接:https://notion.siuze.top/article/944fc3c5-9496-42ad-8306-0b77ee5de0e5
- 声明:本文采用 CC BY-NC-SA 4.0 许可协议,转载请注明出处。
相关文章